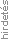

Az SMA többé nem árva betegség!
Kezelhetetlensége miatt a veleszületett gerincvelői izomsorvadás több évtizeden át „árva” betegségnek számított, azonban a betegségmódosító gyógyszerek forgalomba kerülésével a 21. század új perspektívát hozott. Mivel ezen gyógyszerek hatásossága és a terápiakezdés időpontja szorosan összefügg, rendkívül fontos az újszülöttkori szűrővizsgálatokkal történő korai felismerés.
Bevezetés
A veleszületett gerincvelői izomsorvadás (SMA, vagyis spinalis muscularis atrophia) történelmének kezdetei a 19. század végére nyúlnak vissza, Werdnig és Hoffmann ekkor írta le a betegség legsúlyosabb formájának klinikai tüneteit. Az SMA – felismerése ellenére – a 20. század végéig „árva” betegség volt, nem csupán ritkasága, hanem kezelhetetlensége miatt is, hiszen nem álltak rendelkezésünkre olyan terápiás lehetőségek, melyek jó irányba módosították volna a kórkép lefolyását és kimenetelét. A múlt század 50-es éveiben további lépésként felismerték a gyógyíthatatlan betegség enyhébb, későbbi életkorban kezdődő tünetekkel és lassúbb állapotromlással járó típusait. A jelenleg rendelkezésünkre álló, betegségmódosító gyógyszerek kifejlesztésének előfutáraként 1995-ben Judith Melki francia genetikus leírta az SMA pontos géntérképét, megalapozva ezzel a hatékony gyógyszerek kifejlesztését.1 Így juthatott el az orvostudomány újabb történelmi fordulóponthoz: az SMA többé nem „árva” betegség.
Mindezek eredményeként olyan gyógyszerek kerültek kifejlesztésre, melyek a mozgató idegsejtek működését biztosító fehérje (SMN-fehérje, vagyis survival motor neuron fehérje) képződésének elősegítésére irányulnak, az SMN-fehérjeképződést kódoló valamely génszakaszra kifejtve hatásukat (SMN-dependens betegségmódosító terápia). Kutatások folynak egyéb kezelési lehetőségekkel is, melyek az ingerületátvezetést, az SMN-fehérje stabilizálását, az izomtömeg növekedését gátló myostatin inhibícióját célozzák meg, azonban ezek hatásosságát igazoló eredmények még nem születtek.2 Az SMN-dependens gyógyszeres kezeléssel végzett vizsgálatok rávilágítottak a terápia korai életkorban történő elkezdésének fontosságára, és bizonyították, hogy az általuk elérhető életminőség és a terápiakezdet között igen szoros az összefüggés.3-5 Így a betegség tünetmentes újszülött- és fiatal csecsemőkorban történő felismerésének fontossága miatt az orvostudomány újabb kihívás elé került, amelynek megoldásaképpen az SMA újszülöttkori szűrővizsgálatának bevezetése lehetővé tette a kezelés korai elkezdését. Az eredmények várakozást felülmúlóan bizonyították, hogy bár az SMA nem gyógyítható, de hatékonyan kezelhető, a betegek életesélyeit és életminőségét egyaránt javítja.2
A betegségmódosító terápiák hatásmechanizmusa
Az SMA ritka genetikai betegség, ahol a motoros idegsejtek zavartalan működéséhez nélkülözhetetlen fehérjének génhiba miatti elégtelen képződése okozza a motoneuron-pusztulást. Az élő emberi szervezet sejtjeinek „vegykonyhája” a kromoszóma DNS-ének funkciót hordozó szakasza a génállomány, mintegy 20–25 000 fehérje képződésének „szakácskönyve”.6 Az 5q kromoszómán lévő SMN1-génpár (SMN gén, vagyis survival motor neuron gén) a motoneuronok „táplálékául” szolgáló SMN-fehérje képződését kódolja. SMA esetén az 5q kromoszómában hiányzik az SMN1 gén, deléció, ritkán pedig mutáció következtében. Ezért a fehérje motoros idegsejtekben történő képződése elégtelenné válik, azok elpusztulnak, nem tudják eljuttatni az ingerületet az izmokhoz, melyek a beidegzés romlása, megszűnése miatt sorvadásnak indulnak. A legsúlyosabb típusban az izombénulás kiterjedhet a légzőizmokra is. A beteg életét egy másik génpár: az „alvó” – egészséges egyénnél inaktív – SMN2-génpár aktiválódása mentheti meg, melyről az információ átíródása során csak lényegesen kevesebb, mindössze kb.10%-nyi SMN-fehérje képződése valósulhat meg. Ez nem csupán mennyiségileg elégtelen a sejtek számára, de sokkal gyorsabban el is bomló és kevésbé funkcióképes fehérje képződését biztosítja. Így egyre több motoneuront veszít el a beteg, folyamatosan romlik a mozgásképessége és az életminősége (1. ábra).7,8 Attól függően, hogy a másodlagos génpárból hány másolat van a kromoszómában, a betegségnek többféle típusa lehet. Minél nagyobb a kópiaszám, annál enyhébb az izomsorvadás és annál később jelentkeznek a tünetek, illetve annál tovább élnek a betegek (1. táblázat).9
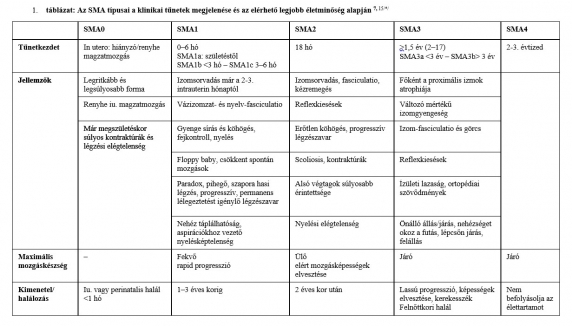
A betegségmódosító gyógyszerek az SMN-fehérje képződését segítik elő az SMN2 génre kifejtett hatás vagy az SMN1 gén pótlása útján. A 21. század nagy vívmányaként ma már három gyógyszer is rendelkezésünkre áll. A megfelelő terápiás hatás eléréséhez nem szükséges mindkét útvonalat egyidejűleg megcéloznunk, hiszen akár az SMN2 gén működését javítjuk, akár a hiányzó SMN1 gént pótoljuk, az eredmény ugyanaz: a motoneuronok számára kellő mennyiségű és minőségű túlélő fehérje képződik folyamatosan, ezáltal pedig biztosítható a még életképes motoros idegsejtek megfelelő működése (2. ábra).
Innovatív betegségmódosító terápiák
A nuszinerszen és a riszdiplám az SMN2 génről történő transzláció során az alternatív hasítás (splicing) befolyásolásával segíti elő a 7-es exon inklúzióját az mRNS transzkripcióba, melynek eredményeként normál funkciójú SMN-fehérje-képződést biztosít. Az elsőként bevezetett gyógyszer a nuszinerszen (Inj. Spinraza 12 mg, Biogen) volt, amelyet 2016-ban vezettek be az Amerikai Egyesült Államokban, majd 2017-ben Európában. 2018 óta hazánkban is több SMA-központ alkalmazza sikeresen.10 A riszdiplám (Evrysdi, Roche) szintén az SMN2 gén pre-mRNS-én fejti ki hatását. Először 2020-ban alkalmazták az USA-ban, 2021-ben pedig európai engedélyt is kapott, és hazánkban is több centrum alkalmazza jó eredményekkel.11 Magának a betegségért felelős hiányzó elsődleges SMN1 génnek a pótlása a génpótló terápia (onaszemnogén abeparvovek: Zolgensma, Novartis), amely rekombináns adenoasszociált 9-es szerotípusú (AAV9) vírusvektorhoz kötve tartalmazza a humán SMN1 gén DNS-ét – ez 2019-től az USA-ban, 2020-tól pedig Európában is engedélyezett terápiás eljárás.12 Mivel szisztémásan kerül beadásra és átjut a vér-agy gáton, mind a motoneuronokban, mind a perifériás szervekben növeli az SMN-fehérje szintjét, az orálisan adott riszdiplámhoz hasonlóan. Nem integrálódik a genomba, nem jut át a mitotikus sejtekbe, így nem változtatja meg a sejtek génállományát.13 Napjainkban ezek a gyógyszeres terápiák egyedi méltányossági elbírálás alapján állami támogatással alkalmazhatók hazánkban.
Nemzetközi tapasztalatok a betegségmódosító terápiákkal
A kezdeti nemzetközi tapasztalatok 2 éves és annál idősebb betegeknél történt alkalmazás eredményeiről számoltak be. Mindhárom gyógyszer alkalmazásánál jótékony hatást figyeltek meg, attól függően, hogy milyen állapotban, mennyi elveszített motoneuron mellett és milyen életkorban kezdődött a kezelés, illetve hogy a betegek az SMA melyik típusában szenvedtek. Az eredmények tovább javultak azoknál a betegeknél, akiknél – bár fennálltak a betegség klinikai tünetei – egyéves életkor előtt elkezdődött a terápia.13,14 A legnagyobb áttörést a legújabb vizsgálatok hozták, amikor még a legtöbb életképes idegsejtjük volt a betegeknek, ezért nem jelentkeztek klinikai tünetek a gyógyszeres kezelés elkezdésekor. Náluk 3–42 napos életkorban, újszülöttként kezdték el valamelyik készítmény alkalmazását. Amikor 1,5-2 év múlva ellenőrző vizsgálatokat végeztek, döntő többségük (még a legsúlyosabb típusban szenvedők) sem igényelték a gépi légzéstámogatást, nem veszítették el nyelésképességüket, sok motoros mérföldkövet elértek az egészséges kortársaikhoz hasonló életkorban. Az eredmény önmagáért beszél, ha felidézzük, hogy a gyógyszerek előtti érában a legsúlyosabb, rapid lefolyású SMA1-ben szenvedő betegeink döntő többségét 2 éves koruk előtt elveszítettük, tartós ágyhoz- és lélegeztetőgéphez kötöttség, folyamatos állapotromlás után (2. táblázat).
2. táblázat: Összesített adatok a nuszinerszen, riszdiplám és onaszenmogén abeparvovek alkalmazásának nemzetközi tapasztalatairól 2–5,10–14/*/
Betegcsoport Tüneteket mutató, 1 évnél fiatalabb betegek Aszimptomatikus SMA-betegek
Terápiakezdeti életkor 3,8–8 hónap 3–42 nap
Terápiakezdet utáni kontrollvizsgálat időpontja 13–24 hó 18–24 hó
Nem igényelt folyamatos gépi lélegeztetést 61–97% 100%
Egyáltalán nem igényelt légzéstámogatást 39–68% 73–100%
Önállóan táplálkozott 68–92% 73–100%
CHOP Intend pontérték ≥40 pont: 71–96% Életkornak megfelelő mérföldkő elérése: 64–100%
Önálló ülés 29–64% 93–100%
Önálló állás 4,5–30% 38–100%
Önálló lépegetés 25–100%
Túlélés 100% 100%
Rövidítések: CHOP Intend (Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders) skála: a mozgásfejlődés mérésére szolgáló pontrendszer a nem ülőképes gyermekeknél. Elérhető maximális értéke 64 pont.
Megjegyzés: A két életkori csoport adatai objektív összehasonlításra nem alkalmasak, mert az SMN2-kópiaszám, a követési idő, valamint a mérési módszerek eltérőek voltak
A terápia tünetmentes életkorban történő elkezdésének korlátai és lehetőségei
A betegség felismerésének időpontja és a gyógyszerekkel elérhető életminőség között bizonyítottan szignifikáns összefüggés áll fenn. A terápia nélküli időablak során ugyanis alattomosan zajlik a már magzati korban megindult motoneuron-pusztulás és csökken a terápia hatásossága. Vagyis az idő a motoneuron! Ezt a kritikus időfaktort a betegség korai felismerésével tudjuk rövidíteni. Sajnos a gyermek megszületésétől a betegség megállapításáig a betegút gyakran hosszú és kanyargós, hiszen az SMA progresszív spektrumbetegség, melynél a tünetek kibontakozása nagymértékben függ az elpusztult motoneuronok arányától. Megszületéskor – ritka kivételtől eltekintve – még az idegsejtek legalább 75%-a életképes, ezért az újszülött nem mutatja a betegség tüneteit.15
A korai diagnózis további nehézsége a nem betegségspecifikus klinikai manifesztáció. Egy beteg kisbaba édesanyja beszámolt arról, hogy „nagyon jó gyerek, soha nem üvölt a kisfiam, legfeljebb csak halkan sírdogál időnként”. Mások elmondták, hogy sokat bukik etetés után és többször mellre kell tenni, mert hamar kifárad a szopásban, így hosszan elnyúlik egy-egy etetési idő, és gyakran visszacsorog az anyatej a szájába. Többen szomorúan említették, hogy gyenge a köhögése, gyakran összegyűlik a légútjaiban a váladék és belázasodik, nehezen-szaporán lélegzik. Előfordult, hogy aggódott az édesanya, mert még 3 hónaposan sem emelte a fejét kisbabája, és kevesebbet mozgott, mint a szomszéd hasonló korú kislánya, de megnyugtatták, hogy vannak lustább kisbabák is. A fenti panaszok egyike sem az SMA kizárólagos tünete. A megszületés után a külvilági léthez alkalmazkodás önmagában is időigényes folyamat. A gyakori légzőszervi fertőzések lehetnek csupán a még éretlen védekező rendszer miatt, a légzőszervi gyulladások SMA nélkül is nehéz-szapora légzéssel járhatnak, a hosszú időt igénylő etetések és bukogatások a még kicsiny befogadó gyomor, nem teljesen működő gyomorszáj-záróizomzat átmeneti, ártalmatlan velejárója, a lustább mozgásfejlődés pedig egyéni sajátosságként is előfordulhat. Azonban ha a tünetek halmozódnak és súlyosbodnak, akkor már felmerül a betegség alapos gyanúja. Viszont amikor már elvész a nyelésképesség és bekövetkezik a légzési elégtelenség, akkorra a motoros idegsejteknek már legalább 90%-a elpusztult, és a gyógyszerek hatása is csökken, néha csupán a további állapotromlás megállításában merül ki.9,15 Ez a visszafordíthatatlan progresszió úgy kerülhető el, hogy nem várunk a klinikai tünetek megjelenésére, hanem már az újszülöttkorban elvégezzük a betegség kimutatását biztosító vizsgálatokat. Az újszülöttkori szűrővizsgálatok éppen azt a célt szolgálják, hogy ezen ritka, de súlyos szövődményekkel, maradandó károsodásokkal, korai gyermekhalállal járó betegségeket már megszületéskor kimutatva, korán elkezdhessük a kezelést, és kivédjük a törvényszerűen súlyos, életre szóló következményeket.16
Az SMA-betegség kockázatának és fennállásának kimutatására többféle laboratóriumi vizsgálat végezhető. SMA esetén a gyermek testi sejtjeinek 5-ös kromoszómáján mindkét génen hiányzik az SMN1-génszakasz. Mivel a betegség autoszom recesszív öröklődésű, a születendő gyermeknél akkor lép fel 25%-os valószínűséggel az SMA, ha az édesanya és az édesapa is hordozza egyik kromoszómáján a génhibát, bár náluk ennek nem lesz következménye a betegség, mivel a génpár másik tagja ép (3. ábra).16 A hibás gén hordozása már a várandósság előtt kimutatható a leendő szülőknél ún. prekoncepcionális egyszeri diagnosztikai vizsgálattal, saját kérésükre. A vizsgálat különösen akkor hasznos, ha a családban született már SMA-beteg gyermek vagy van SMA-beteg családtag. Abban az esetben, ha már várandós az édesanya, magzata megszületése előtt prenatális diagnosztikai vizsgálat történhet, a várandósság 11-12. hetében chorionboholyból vett szövettani mintából, a génhibát hordozó vagy beteg szülőpárnál. A vizsgálat előtt, valamint annak pozitív eredménye esetén a szülők genetikai tanácsadásban részesülnek, melynek során részletes tájékoztatást kapnak a betegség jellemzőiről és a terápiás lehetőségekről.16
Újszülöttkori szűrővizsgálatok
Az újszülöttkori szűrővizsgálatok akkor indokoltak, ha valamely veleszületett betegségnek nincsenek jellemző újszülöttkori tünetei és kezelés nélkül tartós károsodással jár, viszont a korai kezeléssel megelőzhetők a súlyos következmények, valamint bizonyítottan hatásos, életminőséget javító kezelés érhető el a betegek számára. További feltétel a specifikus és érzékeny szűrővizsgálati módszer.16 Az első újszülöttkori szűrővizsgálatot Robert Guthrie vezette be az USA-ban 1961-ben a súlyos szellemi leépüléssel és epilepsziás rohamokkal járó egyik veleszületett anyagcsere-betegség, a fenil-ketonuria kimutatására, szomorú saját családi tragédia kapcsán. Hazánkban 1975-ben kezdődött négy veleszületett anyagcsere-betegség kötelező szűrővizsgálata, melynek palettája 2007-től további 22 anyagcsere-betegségre terjedt ki. Az idén elkezdődött az egyik leggyakoribb örökletes ritka genetikai betegségnek, a mucoviscidosisnak (cisztás fibrózis) a kötelező szűrése is.17
Megismerve az SMA-betegség kóroktanát, kórlefolyását és a gyógyszeres kezeléssel elérhető életminőség-javulást, nem kétséges, hogy hiánytalanul fennállnak az SMA újszülöttkori szűrésének kritériumai. Az SMA újszülöttkori szűrőprogramját az utóbbi években már több országban is elindították (Izrael, Ausztrália, USA, Tajvan, Olaszország, Spanyolország, az Egyesült Királyság, Franciaország, Lengyelország, Szlovénia, Hollandia, Ukrajna, Szerbia), sőt egyes államokban (Németország, Norvégia, Belgium) az összes újszülöttre is kiterjesztették. A szülők körében csaknem teljes a szűrővizsgálat igénybevétele.18,19
A hamarosan megkezdődő újszülöttkori SMA-szűrés kutatási programjával hazánkban is új perspektíva nyílik. Ennek a vizsgálatnak nagy előnye, hogy már akkor megállapítható lesz a betegség fennállása, amikor még a legtöbb életképes, megmenthető motoros idegsejtje van az újszülöttnek, így a fiatal csecsemőknél elkezdett gyógyszeres kezeléssel akár a kortársaihoz hasonló mozgásfejlődés érhető el, elkerülhető a gépi lélegeztetés, a gyomorba vezetett csövön keresztül történő táplálás, valamint a korai tragikus kimenetel. Nem csupán a gyermekek élettartama, hanem életminősége is nagymértékben javulhat. A vizsgálat kutatási programját az Emberi Erőforrások Minisztériuma dolgozta ki, amely biztosítja a szükséges forrásokat is. A program önkéntes, a szűrés igényléséről a szülők dönthetnek. A gyermek számára nem jelent újabb vérvételt, mivel az SMA-szűrővizsgálat is a kötelező anyagcsereszűrésre levett vérből történik. A minták vizsgálatát a Szegedi Tudományegyetem és a Semmelweis Egyetem Anyagcsereszűrő Központja fogja végezni, a program országos koordinációjáért pedig a Magyarországi Református Egyház Bethesda Gyermekkórháza felelős. A kutatási program feladata a vizsgálómódszer megbízhatóságának igazolása, míg végső célja az újszülöttkori kötelező szűrővizsgálatoknak az SMA-szűréssel történő kibővítése minden hazánkban születő újszülöttnél. A szűréssel lehetővé válik, hogy a pozitív szűrővizsgálati eredmény molekuláris genetikai vizsgálattal történő megerősítése és pontosítása után a gyógyszeres kezelés még tünetmentes korai életkorban elkezdhető legyen. Ezáltal élet- és egészségnyereséget, az egyéni, családi és társadalmi terhek csökkentését biztosíthatjuk minden SMA-beteg gyermek számára. Születendő gyermekük érdekében a várandós és a leendő édesanyákat a vizsgálat igénybevételére biztatjuk.
Irodalom:
1. Cure SMA Recognizes Judith Melki on the Anniversary of the Discovery of the SMN Gene. https://www.curesma.org/cure-sma-recognizes-judith-melki-on-the-anniversary-of-the-discovery-of-the-smn-gene/
2. Servais L, Baranello G, Scoto M, et al. Therapeutic interventions for spinal muscular atrophy: preclinical and early clinical development opportunities. Expert Opin Investig Drugs. 2021;30(5):519–527.
3. De Vivo DC, Bertini E, Swoboda KJ, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuro-muscul Disord. 2019;29(11):842–856.
4. Strauss KA, Farrar MA, Swoboda KJ, et al. Onasemnogene Abeparvovec-xioi Gene-Replacement Therapy in Presymptomatic Spinal Muscular Atrophy: SPR1NT Study Update (2384). Neurology. 2020;94(15):2384.
5. Glascock J, Sampson J, Haidet-Phillips A, et al, Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromusc Diseases. 2018;5(2):145–158.
6. Szeberényi J. Precíziós medicina. Medicina Könyvkiadó, Budapest, 2019.
7. Schorling DC, Pechmann A, Kirschner J. Advances in Treatment of Spinal Muscular Atrophy – New Phenotypes, New Challenges, New Implications for Care. J Neuromusc Diseases. 2020;7:1–13. DOI 10.3233/JND-190424.
8. Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromusc Disorders. 2018;28:103–115.
9. Szirmai I. (szerk.). A neurogén izombetegségek. Spinalis muscularis atrophiák. Medicina Könyvkiadó, Budapest, 2011.
10. Spinraza alkalmazási előírás. https://www.ema.europa.eu/en/documents/product-information/spinraza-epar-product-information_hu.pdf
11. Evrysdi alkalmazási előírás. https://www.ema.europa.eu/en/documents/product-information/evrysdi-epar-product-information_hu.pdf
12. Zolgensma alkalmazási előírás. https://www.ema.europa.eu/en/documents/product-information/zolgensma-epar-product-information_hu.pdf
13. Day JW, Mendell JR, Mercuri E, et al. Clinical trial and postmarketing safety of onasemnogene abeparvovec therapy. Drug Safety. 2021;44(10):1109–1119.
14. Mercuri E, Muntoni F, Baranello G, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (STR1VE-EU): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021;20:832–841.
15. Boczán J, Klivényi P, Kálmán B, et al. A Magyar Klinikai Neurogenetikai Társaság konszenzus-ajánlása a felnőttkori spinalis izomatrophia (SMA) kezeléséhez. Ideggyógy Sz. 2021;74(3-4):79–86.
16. Farrar MA, Kiernan MC. The Genetics of Spinal Muscular Atrophy: Progress and Challenges. Neurotherapeutics. 2015;290–302.
17. Szakmai tájékoztató. https://anyagcsere.sed.hu/sites/anyagcsere.sed.hu/files/down/szakmai_tajekoztato2.pdf
18. Kariyawasam DST, Russell JS, Wiley V, et al. The implementation of newborn screening for spinal muscular atrophy: the Australian experience. Genet Med. 2020;22:557–565.
19. Dangouloff T, Vrscaj E, Servais L, et al. The SMA NBS World Study Group: Newborn screening programs for spinal muscular atrophy worldwide Where we stand and where to go- Neuromusc Disorders. 2021;31(6):574–582.
a szerző cikkei
