
Új magyar módszer segítheti a fehérjekutatást
A Nature Communications folyóiratban megjelent tanulmányában a HUN-REN–ELTE Fehérjemodellező Kutatócsoport olyan matematikai módszer alapjait fektette le, amely fehérjék háromdimenziós szerkezetének számítógépes összehasonlítását teszi lehetővé. Az LoCoHD (Local Composition Hellinger Distance) technika az atomok helyzete mellett már az atomok kémiai információját is figyelembe veszi.
Szervezetünkben a fehérjék látják el a sejtek működéséhez szükséges feladatokat, ugyanis molekuláris gépekként képesek többek között egymást ki- és bekapcsolni, a DNS-ről információt átírni, kis- és nagymolekulák szállítását véghezvinni, valamint kémiai reakcióutakat szabályozni. Mindezek sikeréhez azonban elengedhetetlen az, hogy a szóban forgó fehérje a megfelelő téralkatot, tehát a saját 3D elrendeződését felvegye. Az atomok fehérjében történő elrendeződésének meghatározására több kísérleti módszer is rendelkezésre áll (röntgen-krisztallográfia, nukleáris magrezonancia spektroszkópia, krio-elektronmikroszkópia), melyekkel a fehérjekutatók az utóbbi pár évtizedben közel 220 ezer fehérje alakját derítették fel. Ezek az eredmények pedig egyre inkább igénylik azoknak a számítógépes módszereknek az elterjedését és fejlődését, melyek ezeket az elrendeződéseket képesek elemezni.Ilyen módszer az új, LoCoHD névre hallgató algoritmus is, amely a fehérjékben található aminosav-környezetek kémiai jellege (pl. elemösszetétel, töltés, víztaszító jelleg stb.) alapján hasonlít össze téralkatokat. A Perczel András kutatócsoportjában dolgozó Fazekas Zsolt, az ELTE Hevesy György Kémia Doktori Iskola hallgatója által fejlesztett módszer egyszerű, 0 és 1 közötti skálán dönti el, hogy a szóban forgó szerkezetek egymástól mennyire különböznek. A 0 közeli értékek magas hasonlóságot feltételeznek az atomi elrendeződések és kémiai sajátságok között, míg az 1 közeli értékek azt jelzik, hogy az összehasonlított fehérjék merőben más tulajdonságokkal rendelkezhetnek. A kapott számérték (ún. metrika) felhasználásával így újfajta információ nyerhető a tanulmányozott rendszerről.
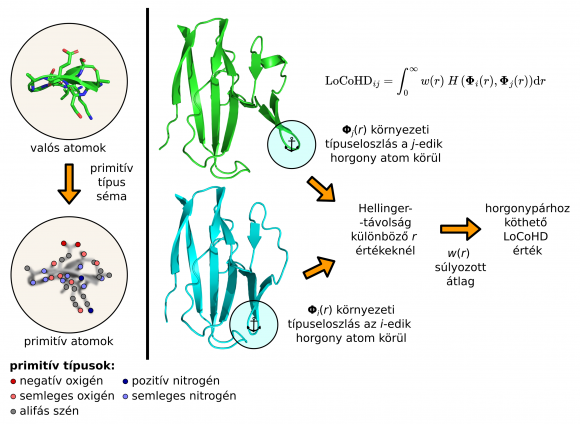
Az algoritmus többlépéses protokoll alapján állítja elő a szerkezeti különbséget jellemző számot. Első lépésben a fehérjék atomjait átalakítja primitív atomokká. Ezeket olyan, virtuálisan felcímkézett atomokként képzelhetjük el, amelyeknek címkéi az adott atom kémiai jellegére utalnak (pl. „pozitív töltésű nitrogén”, „negatív töltésű oxigén”, „semleges töltésű oxigén”, „aromás szén”). A címkék előállítása ún. primitív típus séma alapján történik, ami táblázat formájában jelzi, hogy a valódi atomokat hogyan alakítsuk át primitív atomokká. A felhasználó a táblázatot szabadon kialakíthatja, rögzítve a módszer kémiai felbontását. Második lépésben történik az összehasonlítási pontok meghatározása, amihez kiválasztják a primitív atomok egy részét. A kiválasztott, speciális primitív atomokat horgonyatomoknak nevezik. A horgonyatom-párokhoz az algoritmus összehasonlítást végez, amelynek eredménye a kívánt hasonlóságot adja meg. Ezek a számok kezelhetők a fehérje kisebb részleteinek szintjén, vagy átlagolhatók, és akkor a teljes fehérjét jellemzik.
A Nature Communications folyóiratban megjelent tanulmányban a kutatók kiemelték, hogy a módszer a fehérjekutatásban jól ismert, kétévente megrendezett CASP (Critical Assessment of Protein Structure Prediction) versenyek során is felhasználható. Ezen az eseményen a versenyzők különböző algoritmusokkal igyekeznek a számukra ismeretlen térszerkezetű fehérjék alakjait megbecsülni. Az indulók kiértékelésére a CASP bírák számos szerkezet-összehasonlító módszert alkalmaznak, azonban ezek közül egyik sem veszi figyelembe a létrejövő lokális aminosav-környezetek kémiai jellegét. A 2020-ban megrendezett CASP14 verseny adatait felhasználva most a kutatók több lemodellezett fehérje összehasonlító elemzését is elvégezték, köztük a mesterséges intelligencia alapú AlphaFold2 módszer által jósolt térszerkezetekét is. Ezek közül kiemelték a SARS-CoV-2 vírus ORF8 nevezetű fehérjéjének analízisét. A fehérjéhez tartozó modellszerkezetekben olyan aminosav-környezeteket azonosítottak, amelyek kölcsönhatási mintázataikban jelentősen eltérnek a kísérleti szerkezetben található környezetekkel.
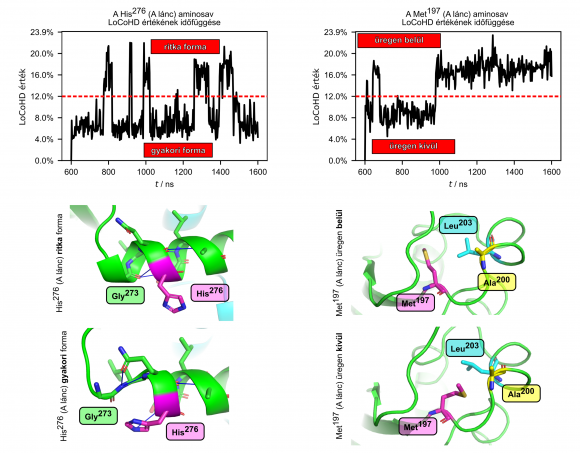
A statikus térszerkezetek tanulmányozása mellett a kutatók azt is megvizsgálták, hogy a módszer alkalmas-e a fehérjék belső mozgásának analízisére. Ehhez molekuláris mozgások tanulmányozására alkalmas szimulációkat, illetve szerkezeti sokaságokból kinyert adatokat használtak fel. Az egyik vizsgált rendszer a podocin fehérje volt, amely létfontosságú feladatokat lát el a vesében, és mutációi súlyos, gyakran halálhoz vezető elváltozásokat képesek okozni. A LoCoHD módszer segítségével a fehérjében olyan aminosavakat azonosítottak, amelyek a podocin mozgása során nagy kémiai környezetváltozásokon esnek át, így befolyásolják annak térszerkezetét és funkcióját.
A technika sikeresen teljesített a HIV-1 kapszidfehérje vizsgálatakor is, ahol a vírus burkának kialakulásában kritikus szerepet betöltő aminosavat emelt ki.
A fehérjeszerkezetek hatékonyabb tanulmányozása nem csak a kutatók számára rendkívül izgalmas, de általa közelebb kerülhetünk a súlyos betegségeket okozó kórokozók jobb megismeréséhez, ezzel a gyógyszerek és a terápiás eljárások fejlesztéséhez is.





















